We develop new tools to infer complex microbial traits from environmentally-derived DNA and apply these tools to large genomic and metagenomic datasets in order to assess the distribution of traits across species and environments. In doing so, we hope to answer questions about (1) what microbes are doing/can do in different habitats, (2) which microbes in a habitat perform particular functions, (3) how certain traits may lead to a fitness benefit/detriment in a particular environmental context, and (4) how complex traits evolve across the tree of life.
In order to accomplish these goals, in addition to developing novel bioinformatics approaches and predictive models to analyze genomic and metagenomic data, we develop dynamical models to construct precise hypotheses about trait evolution and collaborate with experimentalists and field biologists to test these hypotheses.
Below is a sampling of major ongoing projects at the mGAMUT Lab:
Inferring Microbial Traits in Communities to Understand a Changing Climate
TL;DR - We develop computational tools to infer complex microbial traits (i.e., those that can’t be reduced to gene presence/absence) directly from genomic and metagenomic data in order to better represent these microbes in biogeochemical models
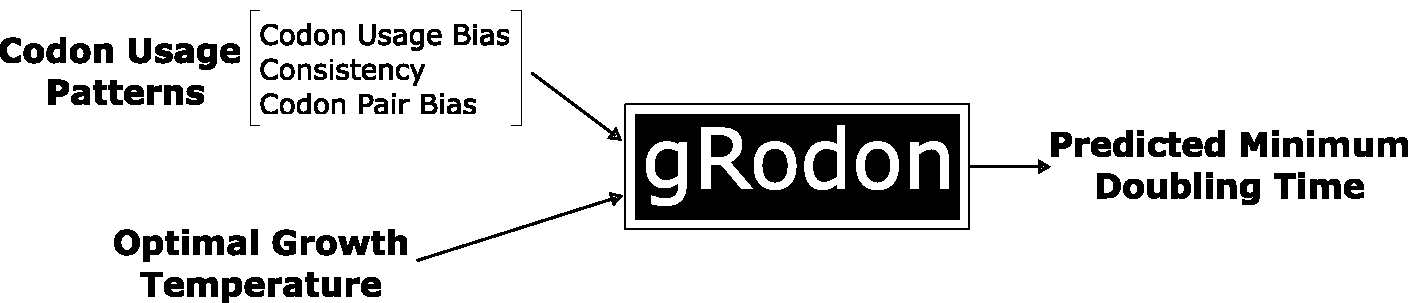
Our research leverages environmental genomic data (“metagenomes”) to untangle the dense networks of interactions by which microbes produce community-level outputs relevant to ecosystem health. Microbes play a critical role in regulating global biogeochemical cycles (i.e., how biologically-relevant elements like carbon move through living and non-living systems) – and a deep understanding of microbial growth at the community scale is essential in order to build accurate models to predict future conditions and design appropriate global mitigation strategies in the face of rapid climate change. While microbial ‘omic datasets are increasingly rich in detail, we lack the tools to link these fine-scale descriptions of community composition to community behavior and outputs. We develop open-source, trait-based frameworks that bridge that gap. For example, we maintain an open-source and user-friendly R package, gRodon, to estimate the maximum growth rates of mixed-species communities directly from environmental metagenomes (see details below). While our research in this area is primarily computational, we work closely with wet-lab collaborators.
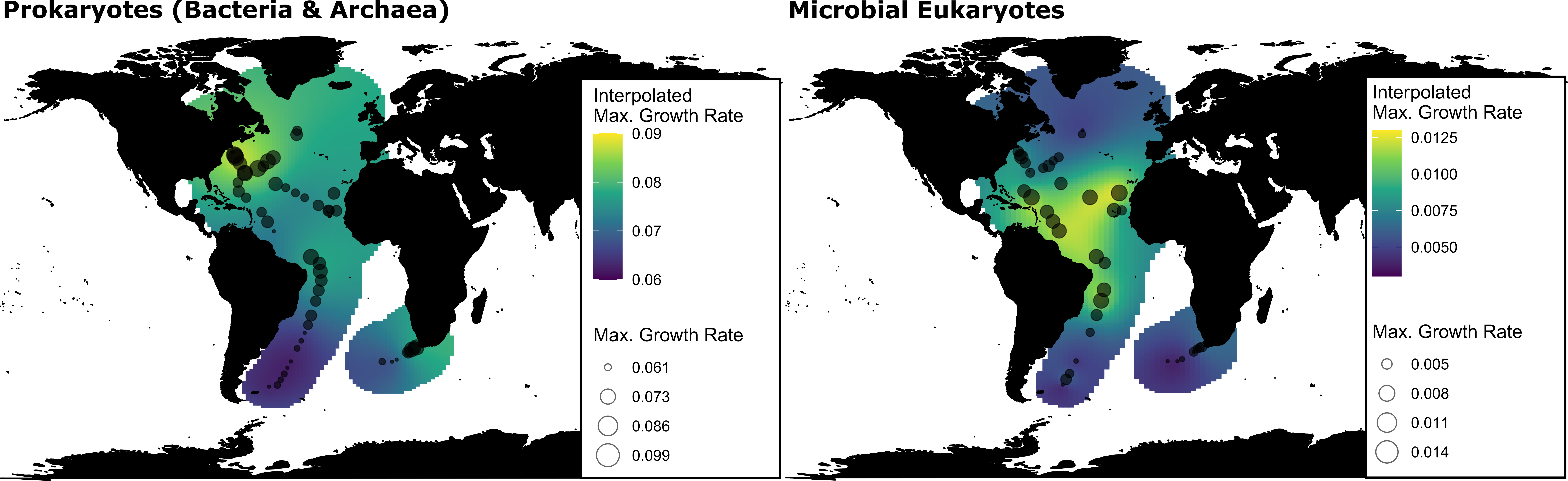
The Maximum Growth Rates of Bacteria, Archaea, and Eukaryotes
TL;DR - We develop computational tools to predict microbial growth phenotypes directly from genomes and metagenomes.
Hey! Does this all seem a bit technical? Check out our article in Frontiers Young Minds for an explanation of how we use DNA to predict how fast bacteria grow that should be accessible to all ages
To-date efforts to functionally annotate metagenomes have largely been concerned with reconstructing the metabolic potential of communities. While microbial metabolism is incredibly diverse and an important aspect of functional ecology, microbes are more than just the metabolic pathways they encode. We have built both genomic predictors and curated trait databases to help characterize the distribution of traits like maximal growth rate in natural communities. For example, we built a novel genomic estimator of maximal growth rate based on codon usage statistics that outperforms previous methods.
In turn, we used this estimator to build a comprehensive database of over 200,000 growth rate estimates from genomes, metagenomes, and single-cell amplified genomes to survey growth potential across the range of prokaryotic diversity. The distribution of growth potentials suggested a natural divide between oligotrophs and coptiotrophs, and led us to propose a redefinition of these broad classes of microbes in terms of their selective environment. Additionally, our database revealed how culture collections, particularly of marine microbes, are strongly biased towards fast-growing organisms, with most environmentally-derived genomes on average having much slower predicted maximal growth rates than cultured isolates, illustrating that the current picture of microbial diversity is not only incomplete, but also highly skewed.
We have implemented our growth-rate estimator in a user-friendly R package called gRodon. More recent developments have allowed us to apply gRodon to both microbial eukaryotes, as well as mixed-species communities of organsisms using metagenomics.
Moving forward, we hope to leverage large phenotypic datasets to build improved genomic predictors for a variety of microbial traits - ultimately building an unbiased picture of microbial form and function in natural environments. Most recently, we have improved our tool’s performance by leveraging phylogenetic information.
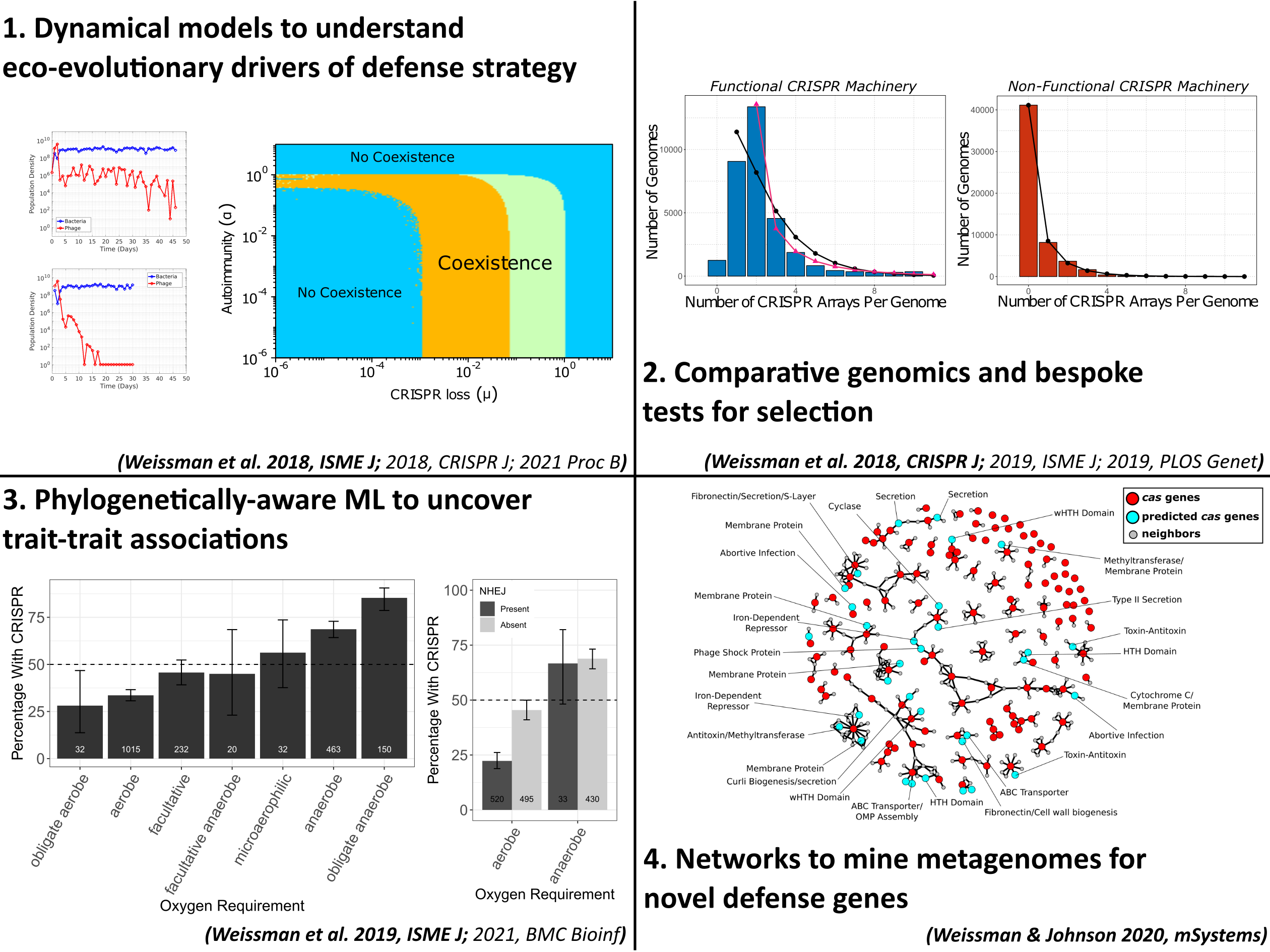
Microbial Antiviral Defense Systems
TL;DR - We combine mathematical models with comparative genomics to better understand the forces shaping the evolution of anti-viral defenses in microbes
Hey! Does this all seem a bit technical? Check out our article in Frontiers Young Minds for an explanation of CRISPR immunity that should be accessible to all ages
Viruses that infect microbes severely impact their hosts’ population and evolutionary dynamics. In an ecological context, these viruses lead to the release of important nutrients back into the environment and play a role in maintaining community-level diversity. In an evolutionary context, viruses drive the evolution of host immune strategy, often leading to iterative co-evolutionary dynamics. In the microbial world these two contexts are not distinct, with demographic and genetic changes occurring at similar rates, making any separation of scales infeasible. This is especially true at the interface of viral-host interactions, where the set of host defense and viral anti-defense strategies is diverse and fast-evolving. Extensive research has profiled the diversity of host and viral communities, but we know relatively little about the distribution of host defense and viral anti-defense strategies across environments.
What drives selection favoring a particular defense strategy? What new defense systems are still waiting to be discovered? How do defense systems coevolve with pathogens over time, and how does this coevolution feed back into the dynamics of host-virus interactions? A central theme of our previous and ongoing work is the application of tools from the fields of machine learning and complex systems science, alongside more traditional approaches from population genetics and theoretical ecology, to understand the ecology and evolution of antiviral defense strategies.
Metagenomics with Community Biologists in an NYC Waterfront Park
Over the past year, we been collaborating with the Billion Oyster Project and GenSpace, a community biology laboratory also based in Sunset Park, to coordinate volunteer-led sampling and processing of a microbial metagenomic time series from waters in and around the Bush Terminal Piers Park. Bush Terminal Park is a quiet community park nestled in an industrial area of Sunset Park, Brooklyn, a historically working-class immigrant neighborhood. This waterfront park, developed on a former brownfield, is subject to storm- and sea level rise-related flooding, is a social and environmental amenity for area residents, and, in combination with efforts to rezone nearby industrial areas for mixed-use development, is impacted by the contested forces of gentrification. Notably, this park is already an active site of ecological research. The Billion Oyster Project, a community organization building artificial oyster reefs across the New York Harbor, has an active community-maintained reef in a lagoon at this site and plans to build another. Our project has been funded by Con Edison and Experiment.com to coordinate sampling of microbial communities living near these reefs (read more here).
